9. Tutorial: Create iML1515 TRBA model¶
The generation of a TRBA model, a thermodynamics-constrained RBA model, can be achieved in a manner analogous to the generation of a TGECKO model. This process can be executed with minimal effort by reusing the XBA, RBA, and TFA configuration files and workflows that have been established in the preceding tutorials.
Peter Schubert, Heinrich-Heine University Duesseldorf, Institute for Computational Cell Biology (Prof. Dr. M. Lercher), January, 2025
Step 1: Initial setup¶
[1]:
import os
import re
import time
from collections import defaultdict
import numpy as np
import pandas as pd
import cobra
from f2xba import XbaModel, TfaModel, RbaModel
from f2xba import RbaOptimization, RbaResults
from f2xba.utils.mapping_utils import load_parameter_file, write_parameter_file
fba_model = 'iML1515'
baseline_model = 'iML1515_RBA'
target_model = 'iML1515_TRBA'
reference_cond = 'Glucose'
# Create media conditions
media_grs = {'Acetate': ['ac', 0.29], 'Glycerol': ['glyc', 0.47], 'Fructose': ['fru', 0.54],
'L-Malate': ['mal__L', 0.55], 'Glucose': ['glc__D', 0.66], 'Glucose 6-Phosphate': ['g6p', 0.78]}
base_medium = ['ca2', 'cbl1', 'cl', 'co2', 'cobalt2', 'cu2', 'fe2', 'fe3', 'h2o', 'h', 'k', 'mg2',
'mn2', 'mobd', 'na1', 'nh4', 'ni2', 'o2', 'pi', 'sel', 'slnt', 'so4', 'tungs', 'zn2']
conditions = {}
exp_grs = {}
for cond, (carbon_sid, exp_gr )in media_grs.items():
conditions[cond] = {f'EX_{sidx}_e': 1000.0 for sidx in base_medium}
conditions[cond][f'EX_{carbon_sid}_e'] = 1000.0
exp_grs[cond] = exp_gr
print(f'{len(conditions)} minimal media conditions created for simulation')
# Load proteomics
fname = os.path.join('data', 'Ecoli_Schmidt_proteomics.xlsx')
with pd.ExcelFile(fname) as xlsx:
df_mpmf = pd.read_excel(xlsx, sheet_name='proteomics', index_col=0)
print(f'{len(df_mpmf)} records of proteomics loaded from {fname}')
min_confidence_level = 43.0
df_mpmf = df_mpmf[df_mpmf['confidence'] > min_confidence_level]
print(f'{len(df_mpmf)} records with confidence level above {min_confidence_level}')
# Metabolite concentrations to be fixed with a margin (Buckstein, 2008)
nt_concs_µmol_per_l = {
'atp_c': 3560, 'adp_c': 116, 'datp_c': 181, 'ctp_c': 325, 'dctp_c': 184,
'gtp_c': 1660, 'gdp_c': 203, 'dgtp_c': 92, 'utp_c': 667, 'udp_c': 54,
'dttp_c': 256, 'ppgpp_c': 113, 'accoa_c': 1390}
factor = 2.0
metabolite_molar_concs = {sid: (µmol_per_l * 1e-6/factor, µmol_per_l*1e-6*factor)
for sid, µmol_per_l in nt_concs_µmol_per_l.items()}
6 minimal media conditions created for simulation
2347 records of proteomics loaded from data/Ecoli_Schmidt_proteomics.xlsx
2232 records with confidence level above 43.0
Step 2: Create TRBA model¶
The generation of a TRBA model is initiated with an XbaModel that has been configured with the XBA configuration file, which was previously utilized for the RBA model. Subsequently, thermodynamics constraints are introduced using the TfaModel. Finally, the RBA configuration data is applied.
[2]:
# Load GEM and extend the model with data from parameters file
xba_model = XbaModel(os.path.join('SBML_models', f'{fba_model}.xml'))
xba_model.configure(os.path.join('data', f'{baseline_model}_xba_parameters.xlsx'))
tfa_model = TfaModel(xba_model)
tfa_model.configure(os.path.join('data', f'{fba_model}_TFA_tfa_parameters.xlsx'))
rba_model = RbaModel(xba_model)
rba_model.configure(os.path.join('data', f'{baseline_model}_rba_parameters.xlsx'))
rba_model.export(os.path.join('SBML_models', f'{target_model}.xml'))
loading: SBML_models/iML1515.xml (last modified: Thu Dec 5 10:03:46 2024)
6 table(s) with parameters loaded from data/iML1515_RBA_xba_parameters.xlsx (Thu Feb 12 15:27:14 2026)
22 gene product(s) removed from reactions (1494 gene products remaining)
23 gene products added to the model (1517 total gene products)
43 constraint ids added to the model (1920 total constraints)
22 variable ids added to the model (2734 total variables)
3 attributes on reaction instances updated
25 reactions with atom imbalances, e.g. [R_PUACGAMS, R_BIOMASS_Ec_iML1515_core_75p37M, R_BIOMASS_Ec_iML1515_WT_75p37M, ...], check XbaModel.atom_imbalances.
24 reactions with charge imbalances, e.g. [R_BIOMASS_Ec_iML1515_core_75p37M, R_BIOMASS_Ec_iML1515_WT_75p37M, R_FMETTRS, ...], check XbaModel.charge_imbalances.
extracting nucleotide sequence from data/ncbi/chromosome_U00096.3_fasta.txt
chromosome : 4641652 nucleotides, 4308 mRNAs, 22 rRNAs, 86 tRNAs
extracting UniProt protein data from data/uniprot_organism_83333.tsv
1517 proteins created
140 cofactors used in proteins could not be mapped (are not considered). These correspond to 18 CHEBI ids, which could be added to the parameter spreadsheet (chebi2sid):
{'49883': '[4Fe-4S] cluster', '60539': 'Mo-bis(molybdopterin guanine dinucleotide)', '30413': 'heme', '190135': '[2Fe-2S] cluster', '21137': '[3Fe-4S] cluster', '24875': 'Fe cation', '25213': 'a metal cation', '60240': 'a divalent metal cation', '87746': 'prenylated FMN', '18408': 'adenosylcob(III)alamin', '23378': 'Cu cation', '30408': 'iron-sulfur cluster', '79027': 'L-topaquinone', '61717': 'heme c', '60342': 'dipyrromethane', '62814': 'heme d cis-diol', '71302': 'Mo-molybdopterin', '28115': 'methylcob(III)alamin'}
479 cofactors mapped to species ids
1224 enzymes added with default stoichiometry
1 table(s) with parameters loaded from data/iML1515_enzyme_composition_updated.xlsx (Thu Feb 12 15:17:11 2026)
1203 enzyme compositions updated from data/iML1515_enzyme_composition_updated.xlsx
2243 reactions catalyzed by 1224 enzymes
default kcat values configured for ['metabolic', 'transporter'] reactions
1 table(s) with parameters loaded from data/iML1515_predicted_fit_GECKO_kcats.xlsx (Thu Feb 12 15:26:06 2026)
5357 kcat values updated from data/iML1515_predicted_fit_GECKO_kcats.xlsx
0 enzymes removed due to missing kcat values
1920 constraints (+43); 2734 variables (+22); 1517 genes (+1); 5 parameters (+0)
>>> BASELINE XBA model configured!
4 table(s) with parameters loaded from data/iML1515_TFA_tfa_parameters.xlsx (Thu Feb 12 16:42:45 2026)
17 thermo data attributes updated
937 metabolites with TD data covering 1545 model species; (375 not supported)
1897 reactions supported by TD data; (500 not supported)
7410 constraints to add
7410 constraint ids added to the model (9330 total constraints)
3794 ∆rG'/∆rG'˚ variables to add
3794 variable ids added to the model (6528 total variables)
2470 forward/reverse use variables to add
2470 variable ids added to the model (8998 total variables)
1528 log concentration variables to add
1528 variable ids added to the model (10526 total variables)
1897 TD reactions split in forward/reverse, 0 opened reverse direction
2470 fwd/rev reactions to couple with flux direction
2470 attributes on reaction instances updated
2 RHS variables to add
2 variable ids added to the model (11101 total variables)
3811 parameters
2 ∆Gr'˚ variables need relaxation.
2 attributes on parameter instances updated
1528 species (930 metabolites) with TD data from total 1920 (1212)
1897 metabolic/transporter reactions with TD data from total 2397
9330 constraints (+7453); 11101 variables (+8389); 1517 genes (+1); 3811 parameters (+3806)
8 table(s) with parameters loaded from data/iML1515_RBA_rba_parameters.xlsx (Thu Feb 12 15:27:14 2026)
2 compartment(s) added
94 gene products added to the model (1611 total gene products)
94 gene product(s) added for process machineries
94 protein(s) created with UniProt information
140 cofactors used in proteins could not be mapped (are not considered). These correspond to 18 CHEBI ids, which could be added to the parameter spreadsheet (chebi2sid):
{'49883': '[4Fe-4S] cluster', '60539': 'Mo-bis(molybdopterin guanine dinucleotide)', '30413': 'heme', '190135': '[2Fe-2S] cluster', '21137': '[3Fe-4S] cluster', '24875': 'Fe cation', '25213': 'a metal cation', '60240': 'a divalent metal cation', '87746': 'prenylated FMN', '18408': 'adenosylcob(III)alamin', '23378': 'Cu cation', '30408': 'iron-sulfur cluster', '79027': 'L-topaquinone', '61717': 'heme c', '60342': 'dipyrromethane', '62814': 'heme d cis-diol', '71302': 'Mo-molybdopterin', '28115': 'methylcob(III)alamin'}
487 cofactor(s) mapped to species ids for added protein
11101 reactions -> 13521 iso-reactions, including pseudo reactions
4 density constraints added
73 targets in 7 target groups
5 targets in 8 target groups
5 compartments
9330 species
13190 reactions
331 medium metabolites (24 > 0.0 mmol/l)
1 macromolecules (4 components) in dna
25 macromolecules (4 components) in rna
1611 macromolecules (48 components) in protein
5 dummy proteins added
3946 enzymes
7 processes and 6 processing maps
5536 functions, 167 aggregates
>>> RBA model created
update XbaModel with RBA parameters
1 attributes on compartment instances updated
0 reactions removed
1642 macromoledules to add
1642 constraint ids added to the model (10972 total constraints)
5391 RBA constraints to add
5391 constraint ids added to the model (16363 total constraints)
5380 fwd/rev reactions to couple with enzyme efficiency constraints
5380 attributes on reaction instances updated
1643 processing reactions to add
1643 variable ids added to the model (15164 total variables)
5703 parameter values calulated based on 1.00 h-1 growth rate and medium
3792 enzyme concentration variables to add
3792 variable ids added to the model (18956 total variables)
7 process machine concentration variables to add
7 variable ids added to the model (18963 total variables)
27 macromolecule concentration target variables to add
27 variable ids added to the model (18990 total variables)
1 metabolites concentration target variable to add
1 variable ids added to the model (18991 total variables)
5 density target and slack variables to add
5 variable ids added to the model (18996 total variables)
3 Flux/variable bounds need to be updated.
3 attributes on reaction instances updated
1 objectives removed
Dummy FBA objective configured: maximize V_TSMC
3828 variables (reactions) require srefs id for initial assignment
3828 attributes on reaction instances updated
5 function definitions added to XBA model
1 parameters removed following cleanup: ['R_ATPM_lower_bound']
XbaModel updated with RBA configuration
model exported to SBML format: SBML_models/iML1515_TRBA.xml
[2]:
True
Step 3. Load and optimize TRBA model (COBRApy)¶
It should be noted that the gurobipy interface, which will be utilized in the forthcoming examples, has been demonstrated to exhibit a substantial increase in processing speed.
[3]:
start = time.time()
fname = os.path.join('SBML_models', f'{target_model}.xml')
rbam = cobra.io.read_sbml_model(fname)
# Load SBML model with RbaOptimization (required for optimization under COBRApy)
ro = RbaOptimization(fname, rbam)
sigma = ro.avg_enz_saturation
importer_km = ro.importer_km
print(f'average saturation level: {sigma}, importer Km: {importer_km} mmol/l')
all_genes = set(ro.m_dict['fbcGeneProducts']['label'].values)
tx_genes, metab_genes = ro.get_tx_metab_genes()
pm_genes = all_genes.difference(tx_genes.union(metab_genes))
print(f'{len(all_genes)} genes: ({len(tx_genes)}) transporter, ({len(metab_genes)}) metabolic, '
f'({len(pm_genes)}) process machines')
# Set nucleotide concentrations (optional)
for sid, (lb, ub) in metabolite_molar_concs.items():
rbam.reactions.get_by_id('V_LC_' + sid).bounds = (np.log10(lb), np.log10(ub))
print(f'{len(metabolite_molar_concs)} metabolite concentrations constrained')
print(f'Duration: {time.time()-start:.1f} s')
Set parameter Username
Set parameter LicenseID to value 2731209
Academic license - for non-commercial use only - expires 2026-10-31
SBML model loaded by sbmlxdf: SBML_models/iML1515_TRBA.xml (Thu Feb 12 16:48:48 2026)
Thermodynamic use variables (V_FU_xxx and V_RU_xxx) as binary
Thermodynamic constraints (C_F[FR]C_xxx, C_G[FR]C_xxx, C_SU_xxx) ≤ 0
3792 enzymes, 7 process machines, 1897 TD reaction constraints
RBA enzyme efficiency constraints configured (C_EF_xxx, C_ER_xxx) ≤ 0
average saturation level: 0.5, importer Km: 1.0 mmol/l
1611 genes: (492) transporter, (1025) metabolic, (94) process machines
13 metabolite concentrations constrained
Duration: 68.2 s
[4]:
start = time.time()
pred_results = {}
for cond, medium in conditions.items():
rel_mmol_per_l = (sigma / (1.0-min(sigma, .99))) * importer_km
ex_sidx2mmol_per_l = {re.sub('EX_', '', ex_ridx): rel_mmol_per_l for ex_ridx in medium}
ex_sidx2mmol_per_l['h_e'] = 100.0 * sigma # H-symport reactions to be not constraint by main metabolite
ro.medium = ex_sidx2mmol_per_l
solution = ro.solve(gr_min=0.0, gr_max=1.2, bisection_tol=1e-3)
if solution.status == 'optimal':
gr = solution.objective_value
pred_results[cond] = solution
print(f'{cond:25s}: pred gr: {gr:.3f} h-1 vs. exp {exp_grs[cond]:.3f}, '
f'diff: {gr - exp_grs[cond]:6.3f}')
else:
print(f'{cond:25s}: INFEASIBLE')
rr = RbaResults(ro, pred_results, df_mpmf)
df_fluxes = rr.collect_fluxes()
df_all_net_fluxes = rr.collect_fluxes(net=True)
metabolic_rids = [rid for rid in list(df_all_net_fluxes.index) if re.match('(PROD)|(DEGR)', rid) is None]
synthesis_rids = [rid for rid in list(df_all_net_fluxes.index) if re.match('(PROD)|(DEGR)', rid)]
df_net_fluxes = df_all_net_fluxes.loc[metabolic_rids]
df_synt_fluxes = df_all_net_fluxes.loc[synthesis_rids]
df_protein_conc = rr.collect_protein_results('µmol_per_gDW')
df_proteins = rr.collect_protein_results('mg_per_gP')
df_enzyme_conc = rr.collect_enzyme_results('µmol_per_gDW')
df_rna_conc = rr.collect_rna_results('µmol_per_gDW')
df_occupancy = rr.collect_density_results()
df_capacity = rr.collect_density_results(capacity=True)
df_species_conc = rr.collect_species_conc()
rr.report_proteomics_correlation(scale='lin')
rr.report_proteomics_correlation(scale='log')
rr.report_protein_levels(reference_cond)
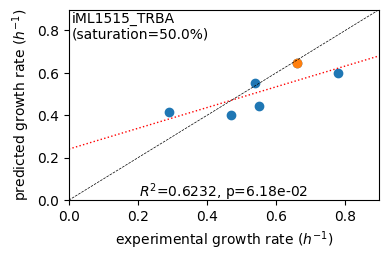
rr.plot_grs(exp_grs, highlight=reference_cond)
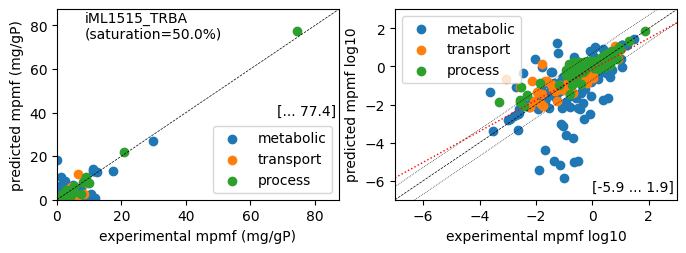
rr.plot_proteins(reference_cond)
rr.save_to_escher(df_net_fluxes[reference_cond], os.path.join('escher', f'{target_model}_TRBA'))
rr.save_to_escher(df_species_conc[reference_cond], os.path.join('escher', f'{target_model}_TRBA'))
print(f'Duration: {time.time()-start:.1f} s')
Acetate : pred gr: 0.414 h-1 vs. exp 0.290, diff: 0.124
Glycerol : pred gr: 0.402 h-1 vs. exp 0.470, diff: -0.068
Fructose : pred gr: 0.553 h-1 vs. exp 0.540, diff: 0.013
L-Malate : pred gr: 0.441 h-1 vs. exp 0.550, diff: -0.109
Glucose : pred gr: 0.646 h-1 vs. exp 0.660, diff: -0.014
Glucose 6-Phosphate : pred gr: 0.597 h-1 vs. exp 0.780, diff: -0.183
Acetate : r² = 0.3685, p = 6.15e-113 (1112 proteins lin scale)
Glycerol : r² = 0.5317, p = 4.47e-185 (1112 proteins lin scale)
Fructose : r² = 0.6779, p = 2.40e-275 (1112 proteins lin scale)
Glucose : r² = 0.8505, p = 0.00e+00 (1112 proteins lin scale)
Acetate : r² = 0.4140, p = 8.21e-51 ( 423 proteins log scale)
Glycerol : r² = 0.5012, p = 2.53e-66 ( 428 proteins log scale)
Fructose : r² = 0.4760, p = 2.26e-59 ( 411 proteins log scale)
Glucose : r² = 0.5040, p = 5.21e-67 ( 429 proteins log scale)
condition: Glucose
1616 proteins in model with total predicted mass fraction of 1000.0 mg/gP
1112 have been measured with mpmf of 807.2 mg/gP vs. 640.4 mg/gP predicted
785 metabolic proteins measured 436.9 mg/gP vs. 330.4 mg/gP predicted
237 transport proteins measured 100.4 mg/gP vs. 46.0 mg/gP predicted
90 processes proteins measured 270.0 mg/gP vs. 264.0 mg/gP predicted
504 proteins not measured vs. 359.6 mg/gP predicted
5 dummy proteins 315.4 mg/gP predicted
499 actual proteins 44.2 mg/gP predicted
total : r² = 0.8505, p = 0.00e+00 (1112 proteins lin scale)
metabolic : r² = 0.5907, p = 4.93e-154 ( 785 proteins lin scale)
transport : r² = 0.4913, p = 2.39e-36 ( 237 proteins lin scale)
processes : r² = 0.9819, p = 1.68e-78 ( 90 proteins lin scale)
total : r² = 0.5040, p = 5.21e-67 ( 429 proteins log scale)
metabolic : r² = 0.4379, p = 2.76e-38 ( 293 proteins log scale)
transport : r² = 0.5831, p = 6.73e-10 ( 46 proteins log scale)
processes : r² = 0.8245, p = 5.18e-35 ( 90 proteins log scale)
1 file(s) exported for "Load reaction data" into Escher maps
1 file(s) exported for "Load metabolite data" into Escher maps
Duration: 1340.7 s
[5]:
# selected species concentrations
df_species_conc.loc[list(metabolite_molar_concs)].head()
[5]:
| name | rank | mean mmol_per_l | stdev | Acetate | Glycerol | Fructose | L-Malate | Glucose | Glucose 6-Phosphate | |
|---|---|---|---|---|---|---|---|---|---|---|
| sid | ||||||||||
| atp_c | ATP C10H12N5O13P3 | 1246 | 3.560000 | 2.757564 | 1.780000 | 7.1200 | 1.780000 | 1.780000 | 7.120 | 1.780000 |
| adp_c | ADP C10H12N5O10P2 | 1301 | 0.174000 | 0.089853 | 0.232000 | 0.0580 | 0.232000 | 0.232000 | 0.058 | 0.232000 |
| datp_c | DATP C10H12N5O12P3 | 1302 | 0.135750 | 0.110839 | 0.090500 | 0.0905 | 0.090500 | 0.090500 | 0.362 | 0.090500 |
| ctp_c | CTP C9H12N3O14P3 | 1288 | 0.379685 | 0.209675 | 0.251642 | 0.6500 | 0.223186 | 0.251642 | 0.650 | 0.251642 |
| dctp_c | DCTP C9H12N3O13P3 | 1306 | 0.092000 | 0.000000 | 0.092000 | 0.0920 | 0.092000 | 0.092000 | 0.092 | 0.092000 |
(Optional) Track progress¶
[6]:
import scipy
import numpy as np
number = 9
xy = np.array([[df_mpmf.at[gene, reference_cond], df_proteins.at[gene, reference_cond]]
for gene in df_proteins.index if gene in df_mpmf.index])
log10_x, log10_y = rr.get_log10_xy(xy)
lin_pearson_r, _ = scipy.stats.pearsonr(xy[:, 0], xy[:, 1])
log_pearson_r, _ = scipy.stats.pearsonr(log10_x, log10_y)
predictions = load_parameter_file('protein_predictions.xlsx')
data = [[number, target_model, lin_pearson_r**2,log_pearson_r**2, len(xy), len(log10_x)]]
cols = ['No', 'model', 'lin r2', 'log r2', 'lin proteins', 'log proteins']
df = pd.DataFrame(data, columns=cols).set_index('No')
if number in predictions[reference_cond].index:
predictions[reference_cond].drop(index=number, inplace=True)
predictions[reference_cond] = pd.concat((predictions[reference_cond], df)).sort_index()
write_parameter_file('protein_predictions.xlsx', predictions)
predictions[reference_cond]
1 table(s) with parameters loaded from protein_predictions.xlsx (Thu Feb 12 16:45:53 2026)
1 table(s) with parameters written to protein_predictions.xlsx
[6]:
| model | lin r2 | log r2 | lin proteins | log proteins | |
|---|---|---|---|---|---|
| No | |||||
| 1 | iML1515_default_GECKO | 0.033244 | 0.190225 | 1018 | 299 |
| 2 | iML1515_modified_GECKO | 0.135981 | 0.201613 | 999 | 322 |
| 3 | iML1515_predicted_GECKO | 0.078803 | 0.268769 | 999 | 308 |
| 4 | iML1515_manual_adjust_GECKO | 0.205413 | 0.333470 | 999 | 308 |
| 5 | iML1515_predicted_fit_GECKO | 0.869850 | 0.549757 | 999 | 313 |
| 6 | iML1515_RBA | 0.855003 | 0.479941 | 1112 | 428 |
| 8 | iML1515_TGECKO | 0.869850 | 0.562074 | 999 | 317 |
| 9 | iML1515_TRBA | 0.850468 | 0.504015 | 1112 | 429 |
(Alternative) gurobipy - model optimization¶
[7]:
# Load TRBA model using gurobipy
start = time.time()
fname = os.path.join('SBML_models', f'{target_model}.xml')
ro = RbaOptimization(fname)
sigma = ro.avg_enz_saturation
importer_km = ro.importer_km
print(f'average saturation level: {sigma}, importer Km: {importer_km} mmol/l')
all_genes = set(ro.m_dict['fbcGeneProducts']['label'].values)
tx_genes, metab_genes = ro.get_tx_metab_genes()
pm_genes = all_genes.difference(tx_genes.union(metab_genes))
print(f'{len(all_genes)} genes: ({len(tx_genes)}) transporter, ({len(metab_genes)}) metabolic, '
f'({len(pm_genes)}) process machines')
# Set nucleotide concentrations (optional)
orig_concs = ro.set_tfa_metab_concentrations(metabolite_molar_concs)
print(f'{len(orig_concs)} metabolite concentrations constrained')
print(f'Duration: {time.time()-start:.1f} s')
SBML model loaded by sbmlxdf: SBML_models/iML1515_TRBA.xml (Thu Feb 12 16:48:48 2026)
MILP Model of iML1515_TFA_RBA
18996 variables, 16363 constraints, 150325 non-zero matrix coefficients
3792 enzymes, 7 process machines, 1897 TD reaction constraints
RBA enzyme efficiency constraints configured (C_EF_xxx, C_ER_xxx) ≤ 0
average saturation level: 0.5, importer Km: 1.0 mmol/l
1611 genes: (492) transporter, (1025) metabolic, (94) process machines
13 metabolite concentrations constrained
Duration: 34.2 s
[8]:
# Optimize model using gurobipy and analyze results
start = time.time()
pred_results = {}
for cond, medium in conditions.items():
rel_mmol_per_l = (sigma / (1.0-min(sigma, .99))) * importer_km
ex_sidx2mmol_per_l = {re.sub('EX_', '', ex_ridx): rel_mmol_per_l for ex_ridx in medium}
ex_sidx2mmol_per_l['h_e'] = 100.0 * sigma # H-symport reactions to be not constraint by main metabolite
ro.medium = ex_sidx2mmol_per_l
solution = ro.solve(gr_min=0.0, gr_max=1.2, bisection_tol=1e-3)
if solution.status == 'optimal':
gr = solution.objective_value
pred_results[cond] = solution
print(f'{cond:25s}: pred gr: {gr:.3f} h-1 vs. exp {exp_grs[cond]:.3f}, '
f'diff: {gr - exp_grs[cond]:6.3f}')
else:
print(f'{cond:25s}: INFEASIBLE')
rr = RbaResults(ro, pred_results, df_mpmf)
df_fluxes = rr.collect_fluxes()
df_all_net_fluxes = rr.collect_fluxes(net=True)
metabolic_rids = [rid for rid in list(df_all_net_fluxes.index) if re.match('(PROD)|(DEGR)', rid) is None]
synthesis_rids = [rid for rid in list(df_all_net_fluxes.index) if re.match('(PROD)|(DEGR)', rid)]
df_net_fluxes = df_all_net_fluxes.loc[metabolic_rids]
df_synt_fluxes = df_all_net_fluxes.loc[synthesis_rids]
df_protein_conc = rr.collect_protein_results('µmol_per_gDW')
df_proteins = rr.collect_protein_results('mg_per_gP')
df_enzyme_conc = rr.collect_enzyme_results('µmol_per_gDW')
df_rna_conc = rr.collect_rna_results('µmol_per_gDW')
df_occupancy = rr.collect_density_results()
df_capacity = rr.collect_density_results(capacity=True)
df_species_conc = rr.collect_species_conc()
rr.report_proteomics_correlation(scale='lin')
rr.report_proteomics_correlation(scale='log')
rr.report_protein_levels(reference_cond)
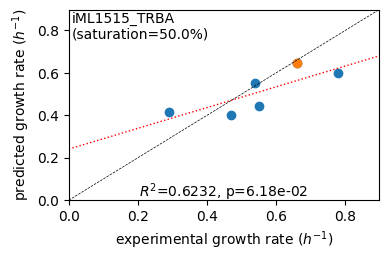
rr.plot_grs(exp_grs, highlight=reference_cond)
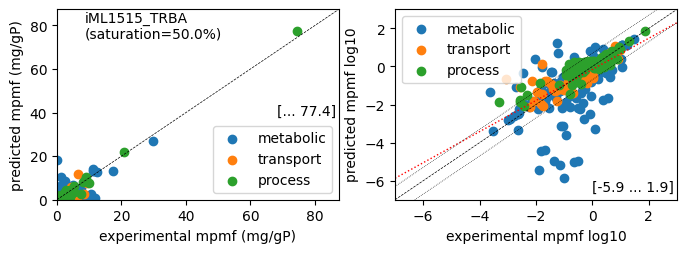
rr.plot_proteins(reference_cond)
rr.save_to_escher(df_net_fluxes[reference_cond], os.path.join('escher', target_model))
rr.save_to_escher(df_species_conc[reference_cond], os.path.join('escher', target_model))
print(f'Duration: {time.time()-start:.1f} s')
Acetate : pred gr: 0.414 h-1 vs. exp 0.290, diff: 0.124
Glycerol : pred gr: 0.402 h-1 vs. exp 0.470, diff: -0.068
Fructose : pred gr: 0.553 h-1 vs. exp 0.540, diff: 0.013
L-Malate : pred gr: 0.441 h-1 vs. exp 0.550, diff: -0.109
Glucose : pred gr: 0.646 h-1 vs. exp 0.660, diff: -0.014
Glucose 6-Phosphate : pred gr: 0.597 h-1 vs. exp 0.780, diff: -0.183
Acetate : r² = 0.3647, p = 1.85e-111 (1112 proteins lin scale)
Glycerol : r² = 0.5304, p = 2.10e-184 (1112 proteins lin scale)
Fructose : r² = 0.6775, p = 4.84e-275 (1112 proteins lin scale)
Glucose : r² = 0.8505, p = 0.00e+00 (1112 proteins lin scale)
Acetate : r² = 0.4254, p = 1.40e-53 ( 431 proteins log scale)
Glycerol : r² = 0.5002, p = 3.04e-65 ( 422 proteins log scale)
Fructose : r² = 0.4623, p = 1.79e-57 ( 414 proteins log scale)
Glucose : r² = 0.4980, p = 4.93e-66 ( 430 proteins log scale)
condition: Glucose
1616 proteins in model with total predicted mass fraction of 1000.0 mg/gP
1112 have been measured with mpmf of 807.2 mg/gP vs. 640.4 mg/gP predicted
785 metabolic proteins measured 436.9 mg/gP vs. 330.4 mg/gP predicted
237 transport proteins measured 100.4 mg/gP vs. 46.0 mg/gP predicted
90 processes proteins measured 270.0 mg/gP vs. 264.0 mg/gP predicted
504 proteins not measured vs. 359.6 mg/gP predicted
5 dummy proteins 315.4 mg/gP predicted
499 actual proteins 44.2 mg/gP predicted
total : r² = 0.8505, p = 0.00e+00 (1112 proteins lin scale)
metabolic : r² = 0.5907, p = 4.92e-154 ( 785 proteins lin scale)
transport : r² = 0.4913, p = 2.39e-36 ( 237 proteins lin scale)
processes : r² = 0.9819, p = 1.68e-78 ( 90 proteins lin scale)
total : r² = 0.4980, p = 4.93e-66 ( 430 proteins log scale)
metabolic : r² = 0.4320, p = 9.59e-38 ( 294 proteins log scale)
transport : r² = 0.5831, p = 6.73e-10 ( 46 proteins log scale)
processes : r² = 0.8245, p = 5.18e-35 ( 90 proteins log scale)
1 file(s) exported for "Load reaction data" into Escher maps
1 file(s) exported for "Load metabolite data" into Escher maps
Duration: 173.9 s